


Rheumatoide Arthritis (rA)
Die Deutsche Gesellschaft für Rheumatologie (DGRh) empfiehlt bei Verdacht auf rheumatoide Arthritis für alle Nicht-Rheumatologen, d. h. insbesondere Hausärzte, hausärztlich tätige Internisten oder Orthopäden zur Annäherung zunächst die folgenden rA-Verdachtskriterien:
- zwei oder mehr geschwollene Gelenke
- Morgensteifigkeit von mehr als einer Stunde und
- erhöhte Blutkörperchensenkungsgeschwindigkeit (BSG) oder erhöhter CRP-Wert gelten hier als Hinweis auf eine mögliche rheumatoide Arthritis
- der Nachweis von Rheumafaktoren oder Autoantikörpern gegen CCP (cyclische citrullinierte Peptide) kann den Verdacht auf eine rheumatoide Arthritis erhärten, aber: ein negativer Befund schließt die Diagnose rA nicht aus
Die definitive Diagnosestellung einer rA sollte dann fachärztlich-rheumatologisch erfolgen. Die bisher hierzu heranzuziehenden und bereits seit 1987 [30] gültigen rA-Klassifikationskriterien der ACR (American College of Rheumatology) wurden 2010 überarbeitet [41]. Die neuen, gemeinsam mit europäischen Wissenschaftlern der EULAR (European League against Rheumatism) erarbeiteten ACR/EULAR-Kriterien stützen sich auf ein Punktesystem. Das Bewertungssystem berücksichtigt die folgenden Variablen:
- A: Anzahl der betroffenen Gelenke (Synovitis)
- B: Rheumaserologie (RF, CCP-Ak)
- C: Entzündungszeichen, akute-Phase-Reaktion (CRP, BSG)
- D: Dauer der Symptome
Tabelle 9: ACR/EULAR-Klassifikationskriterien der rheumatoiden Arthritis [41]
| Kriterium | Punktzahl | |
|---|---|---|
| A | Gelenkbeteiligung (Synovitis) > 10 Gelenke (davon mind. 1 kleines Gelenk) | 5 |
| 4 - 10 kleine Gelenke* (mit/ohne Beteiligung von großen Gelenken) | 3 | |
| 1 - 3 kleine Gelenke* (mit/ohne Beteiligung von großen Gelenken) | 2 | |
| 2 - 10 große Gelenke** | 1 | |
| 1 großes Gelenk** | 0 | |
| B | Serologie (mind. 1 Testergebnis erforderlich) hoch positive RF oder hoch-positive CCP-Ak | 3 |
| niedrig positive RF oder niedrig positive CCP-Ak | 2 | |
| negative RF und negative CCP-Ak | 0 | |
| C | Akute-Phase-Reaktion (mind. 1 Testergebnis erforderlich) erhöhtes CRP oder beschleunigte BSG | 1 |
| unauffälliges CRP und unauffällige BSG | 0 | |
| D | Dauer der Beschwerden ≥ 6 Wochen | 1 |
| < 6 Wochen | 0 |
*:Handwurzel-, Metakarpo-/Metatarsophalangeal- und proximale Interphalangeal-Gelenke
**: Schulter-, Ellenbogen-, Hüft-, Knie-, Sprunggelenk
Patienten mit 6 oder mehr Punkten in diesem Bewertungssystem werden eindeutig als rA-Patienten eingeordnet. Zur Bewertung der Krankheitsaktivität bei Diagnose sowie zum Therapiemonitoring wird häufig der sog. DAS28 (Disease Activity Score 28) herangezogen [42].
Auch wenn aktuell nicht mehr als eigenes Klassifizierungskriterium verwendet, ist die Gelenktopologie dennoch oftmals wegweisend (Abb. 5):
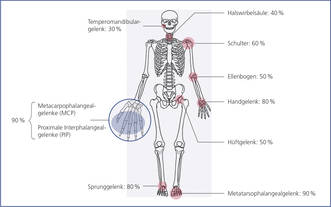
Abbildung 5: Gelenktopologie bei Rheumatoider Arthritis (mit Häufigkeitsangaben). Grundsätzlich kann jedes Gelenk betroffen sein. In 60 - 70 % der Fälle werden kleine Gelenke (Hände und Füße) zuerst befallen. Typisch ist das meist symmetrische Befallsmuster der Polyarthritis.
Die juvenile rheumatoide Arthritis tritt vor dem 16. Lebensjahr auf. Etwa 50 % der Betroffenen entwickeln im Erwachsenenalter eine rheumatoide Arthritis. Eine schwere Verlaufsform der juvenilen chronischen Polyarthritis stellt das Still-Syndrom dar. Sie tritt zumeist bei Kindern und Jugendlichen, gelegentlich auch bei (jungen) Erwachsenen auf. Neben den Symptomen der rheumatoiden Arthritis sind zumeist früh im Krankheitsverlauf auftretende extraartikuläre Organmanifestationen charakteristisch (Fieber, Lymphknotenschwellungen, Hepatosplenomegalie, auch Pericarditis, Pleuritis, Uveitis/Iritis). Der Rheumafaktor ist zumeist negativ, häufiger lassen sich Zellkernantikörper nachweisen [15]. Eine wesentliche Komplikation stellt die Amyloidose dar. Die Prognose der Erkrankung ist ungünstig.
Eine weitere Sonderform der rA, das FELTY-Syndrom, tritt in der Regel erst nach einem längeren Verlauf einer seropositiven chronischen Polyarthritis auf (ca. 3 % der Fälle) und wird vor allem bei schwer verlaufen-den Krankheitsbildern beobachtet. Es ist definiert durch die Symptomkonstellation rheumatoide Arthritis, Leuko-/Thrombopenie und (Hepato-)Splenomegalie. Häufig finden sich Fieberschübe, es besteht eine erhöhte Infektanfälligkeit. Als Folge einer Vaskulitis kann es zu Unterschenkelgeschwüren kommen. In diesen Fällen bedarf es häufig einer Intensivierung der immunsuppressiven Medikation. Ulcera cruris aufgrund anderer Ursachen, z. B. nach vorangegangenen Thrombosen oder bei venöser Insuffizienz, müssen differentialdiagnostisch sorgfältig abgegrenzt werden.
Weitere Kollagenosen
Analog zur rheumatoiden Arthritis kann die Koinzidenz unspezifischer Allgemeinsymptome mit Gelenksymptomen, Hauterscheinungen und anderen Symptomen den Verdacht auf eine andere Autoimmunerkrankung oder Vaskulitis begründen. Häufige mit Kollagenosen assozierte Symptome sind:
- Alopezie
- Schmetterlingserythem, diskoides Erythem
- Photosensibilität
- Orale Ulcerationen
- Arthritis (Gelenkschmerzen)
- Myositis, Muskelschmerzen, Muskelatrophie
- Odem der Hände, RAYNAUD-Phänomen
- Teleangiektasien
- Verhärtung der Haut, Sklerodaktylie
- Mikrostomie, Schluckstörungen
- Sicca-Syndrom
- Pulmonale Fibrose
Frauen sind von Kollagenosen deutlich häufiger betroffen als Männer.
Systematischer Lupus erythematodes (SLE)
| Symptom | Häufigkeit zum Erkrankungsbeginn | Häufigkeit im Verlauf |
|---|---|---|
| Arthritis | 69 % | 84 % |
| Schmetterlingserythem | 40 % | 58 % |
| Fieber | 36 % | 52 % |
| Photosensibilität | 29 % | 45 % |
| RAYNAUD-Phänomen | 18 % | 34 % |
| Serositis | 17 % | 36 % |
| Nephropathie | 16 % | 39 % |
| neurologische Symptome | 12 % | 27 % |
| orale Ulcerosa | 11 % | 24 % |
| Thrombozytopenie | 22 % | |
| Sicca-Syndrom | 16 % |
Die ACR-Diagnosekriterien des systemischen Lupus erythematodes (SLE) umfassen [32]:
- Schmetterlingserythem
- Diskoide Hautveränderungen
- Photosensibilität
- Orale oder nasopharyngeale Ulcerationen (schmerzlos)
- Nichterosive Arthritis, 2 oder mehr periphere Gelenke
- Serositis: Pleuritis oder Perikarditis
- Nierenbeteiligung: Proteinurie > 0,5 g/d oder Erythrozytenzylinder im Harn
- ZNS-Beteiligung: Krampfanfälle oder Psychosen
- Hämolytische Anämie und/oder Leukopenie und/oder Thrombozytopenie*
- Nachweis von anti-dsDNA und/oder anti-Sm (und/oder Anti-Cardiolipin**)
- Nachweis von ANA
*: Leukozyten < 4 G/L, Lymphozyten < 1,5 G/L, Thrombozyten < 100 G/L, mind. zweimal nachgewiesen.
**: mindestens 6 Monate persistierend
Die Diagnose eines SLE gilt bei mehr als 4 erfüllten Kriterien als gesichert.
SJÖGREN-Syndrom
Die europäischen Diagnosekriterien des Sjögren-Syndroms [36] stützen sich auf subjektive und objektivierbare Angaben:
Subjektive Angaben
1. Okuläre Symptome (positive Antwort auf mind. 1 der 3 Fragen):
- > 3 Monate persistierende Probleme wegen trockener Augen?
- Rez. okuläre Missempfindungen (Gefühl von Sand oder Gries im Auge)?
- Benutzung von Tränenersatzflüssigkeit (> 3x täglich)?
2. Orale Symptome (positive Antwort auf mind. 1 der 3 Fragen):
- > 3 Monate persistierendes Gefühl der Mundtrockenheit?
- Rezidivierende oder persistierende Schwellung der Speicheldrüsen als Erwachsener?
- Notwendigkeit von Trinken zum Herunterschlucken trockener Speisen?
Objektive Befunde
3. Okuläre Befunde
- positiver Schirmer-Test* (≤ 5 mm in 5 Minuten), und/oder
- pathologischer Bengalrosatest** (Score > 4 nach van Bijsterfeld)
4. Histopathologie
- fokale lymphozytäre Sialadenitis (Fokusscore*** ≥ 1)
5. Orale Befunde
- pathologisches Szintigramm der Speicheldrüsen, und/oder
- pathologisches Ergebnis der Parotis-Sialographie, und/oder
- unstimulierter Speichelfluss ≤ 1,5 mL/15 min. (Patient < 60 Jahre)
6. Autoantikörper
- Antikörper gegen SS-A/Ro oder SS-B/La, ggf. zusätzlich
- Antinukleäre Antikörper (ANA)
- Rheumafaktor
* Schirmer-Test: Filterpapierstreifen in die untere Konjunktivafalte legen und 5 Minuten belassen. Pathologisch bei < 5 mm Benetzung (Beurteilung unter Beleuchtung)
** van Bijsterveld score: Semiquantitative Bestimmung epithelialer Defekte durch Anfärbung der Binde- und Hornhaut mit Bengalrosa, (maximal 9 Punkte/Auge), pathologisch bei > 4 Punkten
*** Focus: Agglomeration von mind. 50 mononukleären Zellen; Focus-Score = Anzahl Foci pro 4 mm² Drüsengewebe
Für die Diagnose eines primären Sjögren-Syndroms werden > 4 Kriterien (Kriterium 4 und/oder 6 obligat) oder 3 der objektiven Kriterien 3, 4, 5 und 6 gefordert.
Für das skundäre SJÖGREN-Syndrom sind Kriterium 1 oder 2, zusätzlich 2 weitere Kriterien aus 3, 4 und 5 obligat.
Bei SJÖGREN-Syndrom besteht ein erhöhtes Lymphomrisiko. Ausschlusskriterien des SJÖGREN-Syndroms sind Lymphome, AIDS, Sarkoidose, Transplantat-Wirt-Reaktion (GvH-Reaktion), Hepatitis C, Z. n. Strahlentherapie im Kopf-Hals-Bereich, Einnahme von Anticholinergika.
Mischkollagenose (mixed connective tissue disease/MCTD, SHARP-Syndrom)
Die ACR-Diagnosekriterien der Mischkollagenose (MCTD) umfassen Haupt- und Nebenkriterien:
| Hauptkriterien | Nebenkriterien |
|---|---|
| Handödeme* Synovitis (> 1 Gelenk) Myositis (CK, Muskelbiopsie) Raynaud-Phänomen* Akrosklerose* U1-nRNP-Antikörper (obligat) | Alopezie Lekopenie und/oder Anämie und/oder Thrombopenie Trigeminusneuralgie Schmetterlingserythem Pleuritis Perikarditis Arthritis |
Die Diagnose einer MCTD gilt als gesichert bei Anwesenheit von mindestens 4 Hauptkriterien oder 3 Haupt(*)-Kriterien und mindestens 1 Nebenkriterium.
Sklerodermie
Die Klassifikationskriterien der systemischen Sklerodermie [39] umfassen:
| Hauptkriterien | Nebenkriterien |
|---|---|
| Proximale Sklerodermie (symmetrische bilaterale Sklerodermie proximal der Metacarpo bzw. Metatarsophalangeal-Gelenke) | Sklerodaktylie Substanzverlust distaler Finger- bzw. Zehen-Weichteile Ulzerationen an den Fingerkuppen Bilaterale pulmonale Fibrose |
Die Diagnose einer systemischen Sklerodermie gilt bei Erfüllung des Haupt- und mindestens zweier Nebenkriterien als gesichert.
Primäre Vaskulitiden
Immunvaskulitiden weisen eine ähnlich große Symptomenvielfalt wie die Kollagenosen auf. Sie stellen daher eine weitere wichtige Differentialdiagnose im Formenkreis der rheumatologisch-immunologischen Erkrankungen dar. Die einzelnen Formen von Immunvaskulitiden weisen typische Befallsmuster auf und können wie folgt eingeteilt werden:
Tabelle 11: Einteilung der Immunvaskulitiden
| Große Gefäße (Aorta, Aortenabgänge) | Mittelkalibrige Gefäße (größere muskuläre Arterien | Kleinkalibrige Gefäße (kleine Arterien, Arteriolen, Kapillaren, Glomeruli, Venolen) |
|---|---|---|
| Arteriitis temporalis (Riesenzell-Arteriitis) Takayasu-Arteriitis | Polyartheriitis nodosa (PAN) Morbus Kawasaki* Primäre grnulomatöse ZNS-Vaskulitis | Wegener'sche Granulomatose Mikroskopische Polyangitis Churg-Strauss-Syndrom Medikamenten induzierte Vaskulitiden Purpura SCHÖNLEIN-HENOCH Kollagenose assoziierte Vaskulitis Kryoglobulinämische Vaskulitis Postinfektiöse bzw. medikamentöse Immunkomplex-Vaskulitiden M. BEHCET Serumkrankheit |
* = kommt nahezu ausschliesslich bei Kindern vor. Bei den kursiv gedruckten Erkrankungen handelt es sich um Immun-komplex-Vaskulitiden. Als weitere Vaskulitisformen finden sich z. B. paraneoplastische und hypokomplementämische urticarielle Vaskulitiden
Neben der (obligatorischen) Gegenwart unspezifischer Allgemeinsymptome (s. o. Kapitel Weitere Kollagenosen) finden sich bei primärer Vaskulitiden typische Leitsymptome:
- Episkleritis
- Hörsturz, Vertigo
- Hämoptysen
- Mikrohämaturie
- Purpura, Ulcerationen, Livedo reticularis
- Mono-/Polyneritis, Hirninfarkt
- Perimyocarditis, Angina pectoris
- Infarkte: Herz, Niere (Makrohämaturie), Darm
Tabelle 12: Klassifikationskriterien primärer (idiopathischer) Vaskulitiden [40]:
| Morbus WEGENER | CHURG-STRAUSS-Syndrom (CSS) | Polyarteriitis nodosa |
|---|---|---|
| Entzündung in Nase oder Mund (ulzerierend, hämorrhagisch, purulent) | Asthma bronchiale | Gewichtsverlust > 4 kg seit Krankheitsbeginn |
| Lungeninfiltration (radiol.: Rundherde, Kavernen, "fixe" Infiltrationen) | Lungeninfiltration (migratorisch, "flüchtig") | Livedo reticularis |
| Nepritisches Sediment (Erythrozyturie, Ery-Zylinder) | Paranasale Sinusauffälligkeit | unerklärter Hodenschmerz oder Schwellung |
| Eosinophilie (> 10 %) | Myalgien (v. a. untere Extremität) | |
| Mono-/Polyneuropathie | Monneuritis/Polyneuropathie | |
| Histol.: granulomatöse Entzündung (Gefäßwand, peri-/extravaskulär) | Histol.: extravskuläre Eosinophilenakkumulation | Histol.: entzündliche polymorphkernige Gefäßwandinfiltrate |
| distaler Blutdruck > 90 mmHg | ||
| Serum-Kreatinin > 1,5 mg/dL | ||
| HBs-Ag-Nachweis im Serum | ||
| pathologisches Arteriogramm, Aneurysmata, Verschlüsse | ||
| bei 2 von 4 Kriterien: Sensitivität 88 % Spezifität 92 % | bei 4 von 6 Kriterien: Sensitivität 85 % Spezifität 99 % | bei 3 von 10 Kriterien: Sensitivität 82 % Spezifität 87 % |
Reaktive Arthritis und andere seronegative Spondylarthropathien
Reaktive Arthritiden stellen bakteriell induzierte Gelenkerkrankungen dar, die, nach einer auslösenden, primär gelenkfernen Infektion im Urogenital-, Intestinal- oder Respirationstrakt, mit einer Latenzzeit von wenigen Tagen bis Wochen auftreten. Definitionsgemäß lässt sich der Erreger aus der Synovialflüssigkeit nicht kultivieren. Reaktive Arthritiden manifestieren sich vor allem an der unteren Extremität, insbesondere am Sprung- und Kniegelenk. Eine spezielle Untergruppe der reaktiven Arthritis stellt das Reiter-Syndrom (Mono-/Oligoarthritis, Urethritis, Konjunktivitis) dar [9, 19].
Zur Diagnostik können neben der Basisanalytik serologische Untersuchungen herangezogen werden, die Erregerauswahl sollte sich entsprechend der aktuellen klinischen Infektionsanamnese richten (vgl. auch Anhang „Rheumatologische Stufendiagnostik“, 2. und 3. Stufe). Grundsätzlich sollte bei akuter Monarthritis zur Abklärung einer (auch subklinischen!) Urethritis der Chlamydien-Direktnachweis erfolgen. Bei broncho-pulmonalen und gastrointestinalen Infektionen ist dagegen zum Zeitpunkt der Gelenkmanifestation der direkte Erregernachweis am ursprünglichen Ort zumeist nicht mehr möglich. Diagnostisch wegweisend sind der Nachweis erregerspezifischer IgA- und IgM-Antikörper und/oder ein signifikanter Titeranstieg um mindestens 2 Stufen innerhalb von 10 - 14 Tagen. Die Gegenwart von HLA B 27 prädisponiert bei reaktiven Arthritiden zu schweren Krankheitsverläufen.
International akzeptierte Diagnosekriterien der Reaktiven Arthritis sind noch nicht erarbeitet. Von mehreren Gremien existieren Vorschläge:
Kriterien:
- Typischer Gelenkbefall (peripher, asymmetrisch, oligoartikulär, häufig Knie-, Sprunggelenke)
- Typische Anamnese (Diarrhoe, Urethritis) bzw. Manifestation der Infektion an der Eintrittspforte
- Erregerdirektnachweis an der Eintrittspforte (z. B. Urethralabstrich auf Chlamydien)
- Nachweis spezifischer IgA- und/oder IgM-Antikörper bzw. eines signifikanten Titeranstieges
- Vorliegen des HLA B 27-Antigens
- Erregernachweis mittels PCR oder monoklonaler Antikörper
Eine sichere reaktive Arthritis liegt vor bei den Kriterien 1 plus 3 oder 4 oder 6.
Eine wahrscheinliche reaktive Arthritis besteht bei den Kriterien 1 plus 2 und/oder plus 5.
Eine mögliche reaktive Arthritis wird bei Vorliegen des Kriteriums 1 angenommen.
Bei Spondylitis anykylosans (Morbus BECHTEREW) ist der entzündliche Rückenschmerz mit Befall des Ileosakralgelenks typisch. Eine periphere Mon- oder Oligoarthritis vorzugsweise der großen Gelenke der unteren Extremität wird bei 20 – 25 % der Patienten mit Morbus BECHTEREW beobachtet. In 30 % der Fälle tritt sie als Erstsymptom vor den Rückenschmerzen auf [9]. Der Nachweis von HLA B 27 ist bei der Spondylitis ankylosans pathognomonisch (> 95 %, Normalbevölkerung: ca. 8 %), er zählt zusammen mit der beschleunigten BSG zu den Frühdiagnosekriterien [22].
Unter dem Begriff „seronegative Spondarthritiden“ werden - neben den Reaktiven Arthritiden und der Spondylitis ankylosans - vor allem die Arthritis psoriatica sowie Arthritiden bei Morbus Crohn und Colitis ulcerosa subsumiert. Diese Erkrankungen weisen als klinische Gemeinsamkeit ein Syndrom aus Sakroiliitis - Spondylitis - Arthritis auf. Gegenüber der chronischen Polyarthritis lassen sie sich durch das Fehlen von Rheumafaktoren und Rheumaknoten abgrenzen.
Folgende gemeinsame Merkmale charakterisieren die Spondarthritiden:
- Periphere Arthritis, meist asymmetrisch und besonders an den unteren Extremitäten
- Befall von Iliosakralgelenken (Sakroiliitis) und Wirbelsäule (Spondylitis, Syndesmophyten)
- Entzündliche Veränderungen der Insertionen von Sehnen und Bändern (Enthesopathien)
- extraartikuläre Manifestationen (Iridocyclitis, Konjunktivitis, selten Episkleritis, Stomatitis aphthosa, Urethritis, Psoriasis, Erythema nodosum, Pyoderma gangraenosum)
- Familiäre Häufung und hohe Assoziation mit HLA B 27
Entsprechend den Kriterien der European Spondylarthropathy Study Group (ESSG) erfordert die Diagnose einer Spondarthritis die Gegenwart eines entzündlichen Wirbelsäulenschmerzes und/oder einer asymmetrischen Arthritis vorwiegend der unteren Extremität. Zusätzlich muss ein weiteres Kriterium erfüllt sein:
ESSG - Kriterien der Spondylarthropathien
Hauptkriterien:
- entzündlicher Wirbelsäulenschmerz und/oder asymmetrische Arthritis bzw. Arthritis der unteren Extremitäten
Nebenkriterien:
- positive Familienanamnese für Spondylitis ankylosans, Psoriasis, reaktive Arthritis, M. Crohn oder Colitis ulcerosa
- Befund oder Anamnese einer Psoriasis
- M. Crohn oder Colitis ulcerosa
- beidseits wechselnde Gesäßschmerzen
Die Sensitivität der ESSG - Kriterien beträgt 86 (79 - 100) % und die Spezifität 87 %. Auch ohne Röntgenbefund einer Sakroiliitis wird allein mit den anamnestischen und klinischen Daten noch eine Sensitivität von 77 % und eine Spezifität von 89 % erreicht. Das HLA B 27 wurde nicht in den Kriterienkatalog aufgenommen, da es die diagnostische Wertigkeit nicht steigert.
Als diagnostisch wichtige extraartikuläre Krankheitssymptome der Spondylarthropathien gelten vor allem gastrointestinale und urogenitale Symptome sowie Haut- und Schleimhautveränderungen. Andere viszerale Beteiligungen, z. B. kardiale Manifestationen (Aorteninsuffizienz, Myokarditis, Perikarditis, AV-Überleitungsstörungen), unspezifische Begleithepatitiden, Myositis oder Amyloidose, sind sehr selten. Die Assoziation von Spondylarthropathien mit HLA B 27 ist mit ca. 40 (bei isoliert peripher-artikulären Formen) bis 85 % (bei Nachweis einer röntgenologischen Sakroiliitis und anderer axialer Symptome) niedriger als bei der idiopathischen Spondylitis ankylosans.
Kristallarthropathien
Gicht, Chondrokalzinose, Hydroxylapatit-Erkrankung und Hämochromatose-Arthropathie können dieser Gruppe zugeordnet werden.
Gicht
Die Gicht ist Folge einer entzündlichen Reaktion auf die Bildung von Uratkristallen bei bestehender Hyperurikämie. Das klinische Spektrum der Gelenkmanifestationen reicht von der akuten Kristallsynovialitis bis zur chronischen Arthropathie. Die Hyperurikämie kann durch eine renale Ausscheidungsstörung oder endogene Mehrproduktion bedingt sein.
Tabelle 13 Gicht - Begriffsdefinitionen
| Gicht | Störung des Purinstoffwechsels als Allgemeinerkrankung |
|---|---|
| Arthritis urica | Gelenkentzündung, die durch Harnsäurekristalle verursacht wird |
| Gichtsyndrom | Gesamtheit der durch eine Hyperurikämie bedingten (Stoffwechsel-)Veränderungen |
| Hyperurikämie | Erhöhung der Harnsäurewerte über die Löslichkeitsgrenze im Plasma (Normalwert Frauen < 6,2 mg/100 mL = 360 µmol/L, Männer < 7,4 mg/100 mL = 420 µmol/L) |
Folgende Kennzeichen machen die Diagnose der Arthritis urica wahrscheinlich: Monoartikulärer Befall, Akuität (Beschwerdemaximum innerhalb weniger Stunden erreicht), Spontanremission (zumeist Rückbildung innerhalb von 1 bis 2 Wochen). Die Diagnose kann als gesichert gelten, wenn zusätzlich mehrfach erhöhte Harnsäurewerte nachgewiesen werden oder eine prompte Colchicin-Wirkung eintritt.
Die Diagnose der Arthritis urica gilt als sicher bei:
- Nachweis von Uratkristallen in der Synovialflüssigkeit oder Uratablagerung in Geweben
- Bei Vorhandensein von zwei oder mehr der folgenden Kriterien:
- eindeutiger Anamnese bzw. Beobachtung einer Podagra
- eindeutiger Anamnese bzw. Beobachtung von mind. 2 typischen Anfällen an Extremitätengelenken
- eindeutiger Anamnese bzw. Beobachtung einer prompten Reaktion auf Colchicin, d. h. Verminderung der objektiven Entzündungszeichen innerhalb von 48 h nach Gabe
- klinisch nachgewiesener Tophi
Die Hyperurikämie wird hier nicht als Kriterium geführt. Dies bedeutet, dass bei Anwendung der Kriterien die Diagnose nicht schon nach dem ersten Anfall gestellt werden kann, wenn nicht die Großzehe befallen ist (was im Erstanfall nur bei 60 % vorkommt) und mit Colchicin behandelt wurde.
Zusätzliche diagnostische Regeln:
- Bei der Erstattacke ist in > 80 % die untere Extremität betroffen (Podagra 60 %, Sprunggelenk 14 %, Knie 6 %, Fußweichteile/übrige Zehen 2 %), bei je ca. 5 % Ellenbogen-, Hand- und Fingergelenke.
- Eine akute Arthritis an der unteren Extremität beim Mann sollte bis zum Beweis des Gegenteiles als Ver-dachtsfall einer Arthritis urica betrachtet werden.
- Bei Frauen kommt eine Arthritis urica vor der Menopause nur extrem selten vor.
- Die statistische Wahrscheinlichkeit für einen Gichtanfall nimmt mit der Höhe des Harnsäurespiegels zu und beträgt bei Werten von über 9 mg/100 mL etwa 90 %. Die Hyperurikämie ist im Gichtanfall jedoch nicht obligat (mindestens 5 % der Patienten mit Normalwerten!).
- Bei längerbestehenden Ergüssen können sich Uratkristalle auflösen, ein negativer Kristallbefund im Gelenkpunktat schließt die Diagnose der Arthritis urica daher ebenfalls nicht völlig aus.
- Auslösende Ursachen (reichliche Mahlzeiten, Alkoholgenuss, Überanstrengungen, Traumen[19]) sind eher selten zu eruieren; anfallsauslösende Medikamenten sind vor allem Saluretika, Urikosurika (in der Therapieeinleitungsphase) und Penicillin.
- Tophi (Prädilektionsorte: Ohren, Ellenbogen, Füße, Hände) sind im Erstanfall nur sehr selten nach-weisbar (bei über 20jährigem Krankheitsverlauf: 70 %). Die mittlere Latenzzeit beträgt, wie die der ersten röntgenologischen Knochen- und Weichteilveränderungen, 5 bis 7 Jahre.
- Bei Krankheitsbildern mit vermehrtem Umsatz von Nukleoproteiden (Polyzythämien, Leukämien, Psoriasis), bei chronischer Nephropathie und Bleiintoxikation ist an eine sekundäre Gicht zu denken.
Sonstige Symptome und Begleiterkrankungen
Bei Gicht sind folgende Begleiterkrankungen häufig und sollten diagnostisch berücksichtigt werden:
- Übergewicht (bei 75 % der Fälle mindestens + 25 % des Normgewichtes)
- Nephrolithiasis (7 bis 20 %)
- Nephropathie (bei langdauernden Verläufen klinisch bis 50 %, autoptisch häufiger)
- Diabetes mellitus (manifest 10 bis 25 %)
- Hyperlipoproteinämie (über 40 %)
- Leberveränderungen (Fettleber über 60 %)
- Hochdruck, frühzeitige Arteriosklerose (über 40 %)
Chondrokalzinose (Calciumpyrophosphatdihydrat (CPPD)-Kristallarthropathie, Pseudogicht
Der Begriff der Chondrokalzinose beinhaltet die klinisch und anatomisch nachweisbare Verkalkung des Gelenkknorpels. Die Calciumpyrophosphatdihydrat-Kristallarthropathie umfasst alle klinischen Manifestati-onen, die mit der intraartikulären Kristallablagerung verbunden sind. Das Krankheitsbild tritt in 3 Formen auf:
- sporadisch-idiopathisch
- sekundär (u. a. Hyperparathyreoidismus, Hämochromatose, Hypothyreose, Amyloidose, Gicht, Hypomagnesiämie, Hypophosphatasie); auslösend wirkt oft ein Trauma
- hereditär (Endemiegebiete vor allem Slowakei, Chile, Niederlande)
Die Klinik ist sehr variabel, die wichtigsten Formen sind in Tabelle 14 dargestellt.
Tabelle 14: Verlaufsformen und Häufigkeiten der artikulären Chondrokalzinose
| Verlaufsform | Häufigkeit [%] | Klinik |
|---|---|---|
| Pseudo-Arthrose | 50 | wie Gonarthrosen oder Arthrose anderer Lokalisation; je 50 % mit oder ohne akute Entzündungsschübe |
| Pseudogicht | 25 | akute/subakute Mon- bzw. Oligoarthritis; oft Beginn in einem "Muttergelenk" (50 % Knie) und Übergang auf "Tochtergelenke" |
| Asymptomatisch | 20 | röntgenologischer Zufallsbefund in zunehmender Frequenz bei Personen über 55 Jahren |
| Pseude-rA | 5 | akute oder chronische Polyarthritis mit Morgensteifigkeit |
| Pseudo -Neuropathie -Septikämie -Meningitis -Spondylodiszitis | je 1 | rasche Destruktion von Knochenpartien ohne neurologische Defekte Verlauf mit Fieber und Leukozytose Nackensteife, Fieber, akutes Zervikalsyndrom bei Lokalisation in anderen Wirbelsäulenabschnitten |
Laborparameter besitzen keinen diagnostischen Wert. Wichtig ist der Ausschluss von Primärerkrankungen, die als Ursache sekundärer Formen in Frage kommen. Die Diagnose der Chondrokalzinose wird radiologisch oder über den Kristallbefund gestellt. Im Röntgenbild finden sich im typischen Fall lineare und punktierte, "monstranzartige" Verschattungen parallel zur Knorpeloberfläche (v. a. Knie/Ellenbogen) sowie gröbere schollige oder streifige Ablagerungen (Menisken, Disci articulares, Anulus fibrosus der Disci intervertebrales, Synovialmembranen, fibröse Gelenkkapsel, in Sehnen und Bändern).
Das wechselnde klinische Bild der Chondrokalzinose ergibt die Notwendigkeit, bei jeder diagnostisch unklaren Arthropathie nach Gelenkerguss zu fahnden, ihn abzupunktieren und polarisationsoptisch auf Kristalle zu untersuchen. Die sichere Identifizierung eines CPPD-Kristalles beweist die Diagnose, dies gelingt aber nicht in jedem Fall. Zur Unterscheidung von den Uratkristallen der Gicht siehe Tabelle 15:
Tabelle 15: Differenzierung zwischen Urat und CPPD Kristallen
| Uratkristalle | CPPD-Kristalle | |
| Form | nadelförmige, spitze/abgerundete Stäbchen | Rhomben, Stäbchen mit rechteckigen Rändern |
| Größe | meist > 10 nm | meist < 5 nm |
| Polarisation | starke negative Doppelbrechung | schwach positive Doppelbrechung |
Die Abgrenzung der Chondrokalzinose von der Arthritis urica erfolgt neben dem Kristallbefund durch die längere Anfallsdauer von 3 bis 4 Wochen oder den chronischen Verlauf, den vorwiegenden Befall größerer Gelenke und die verzögerte Reaktion auf Colchicin.
Hämochromatose-Arthropathie
Bei rund 64 % der Hämochromatosepatienten kommt es zu einer Hämochromatose-Arthropathie. Hierbei sind die Gelenkveränderungen zumeist eher degenerativer Natur. Es kommen jedoch auch entzündliche Verläufe im klinischen Erscheinungsbild ähnlich einer rheumatoiden Arthritis vor. Charakteristisch sind Schwellung, Auftreibung und Schmerzhaftigkeit der Fingergrundgelenke von Zeige- und Mittelfinger, häufig sind auch Hüft- und Kniegelenk, seltener andere Gelenke betroffen.